|
[O-])Cl](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=c1ccc%28c%28c1%29%5BN%2B%5D%28%3DO%29%5BO-%5D%29Cl&width=200&height=125&arrowdesc=&extraImageSetting=amap)
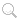
![C[O-].[Na+]](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=C%5BO-%5D.%5BNa%2B%5D&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
 |
[O-]](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=COc1ccccc1%5BN%2B%5D%28%3DO%29%5BO-%5D&width=200&height=125&arrowdesc=&extraImageSetting=amap)
Note: SnAr (nucleophilic aromatic substitution) against an aryl halide. Note that this is neither an Sn2 or Sn1 mechanism. Refer to the [Mechanism] link for more details on how this proceeds via an anionic intermediate that is stabilized by the electron withdrawing nitro group.
|
|
|
[O-])Cl](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=c1cc%28ccc1%5BN%2B%5D%28%3DO%29%5BO-%5D%29Cl&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
[O-]](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=COc1ccc%28cc1%29%5BN%2B%5D%28%3DO%29%5BO-%5D&width=200&height=125&arrowdesc=&extraImageSetting=amap)
Note: SnAr reaction with the electron withdrawing nitro group para to the halide leaving group.
|
|
|
[O-]](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=c1cc%28cc%28c1%29Cl%29%5BN%2B%5D%28%3DO%29%5BO-%5D&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
Warning: SnAr reactions require an electron withdrawing group to be ortho or para to the halide leaving group to stabilize the anionic intermediate. This will not be effective if the groups are meta to each other.
|
|
|
|
|
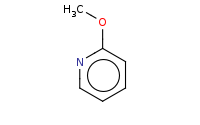
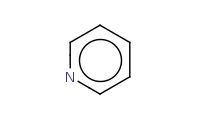
Note: Pyridines react similarly to benzenes with a 'built-in' deactivating group. As a result, they are poor substrates for EAS reactions, but can be quite effective for SnAr (nucleophilic aromatic substitution) reactions.
|
|
|
|
|
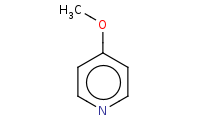
Note: SnAr reaction of pyridine at the para position.
|
|
|
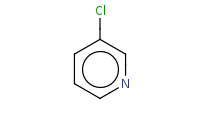
|
|
Warning: SnAr reaction of pyridine only occurs at the ortho and para positions, not meta.
|
|
|
![[Li]C=C](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=%5BLi%5DC%3DC&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
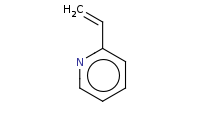
Caution: Very strong nucleophiles, like organolithiums, can substitute onto pyridine rings in an SnAr like reaction even in the absence of a decent leaving group. In this example of a Chichibabin-like reaction, the leaving group is actually a hydride ion.
|
|
|
![[CH2-]c1ccccn1.[Na+]](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=%5BCH2-%5Dc1ccccn1.%5BNa%2B%5D&width=200&height=125&arrowdesc=&extraImageSetting=amap)
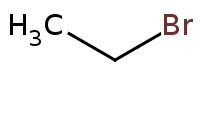
|
|
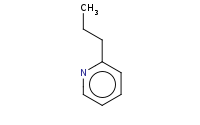
Note: Deprotonated pyridine derivatives behave analogously to enolates, including the capacity for alkylation.
|
|
|
![CC#[C-].[Na+]](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=CC%23%5BC-%5D.%5BNa%2B%5D&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
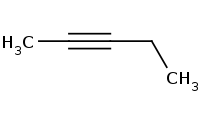
Note: Acetylide ions are effective as strong nucleophiles
|
|
|
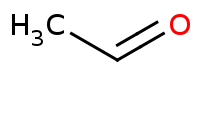
![CC(=C)[Mg]Br](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=CC%28%3DC%29%5BMg%5DBr&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
C)O](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=C%5BC%40H%5D%28C%28%3DC%29C%29O&width=200&height=125&arrowdesc=&extraImageSetting=amap)
Note: Organometallic addition to an aldehyde as a carbon nucleophile. Aqueous workup is assumed to follow and protonate any remaining anions
C)O](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=C%5BC%40%40H%5D%28C%28%3DC%29C%29O&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
|
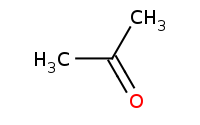
|
|
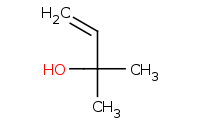
Note: Organometallic addition to a ketone as a carbon nucleophile. Aqueous workup is assumed to follow and protonate any remaining anions
|
|
|
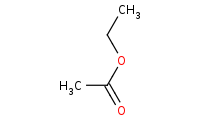
|
|
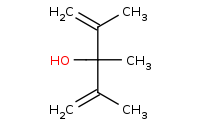
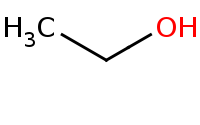
Note: Organometallic addition to a carboxylic acid derivative such as an ester. After the first addition, the C-O bond can break with an alkoxide leaving group (feasible when using a very strong nucleophile like an organometallic). The resulting ketone is even more reactive than the original ester, thus a second equivalent of organometallic will add, yielding a tertiary alcohol. Refer to the [Mechanism] link for more details.
|
|
|
|
|
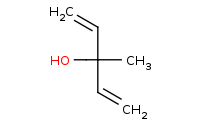
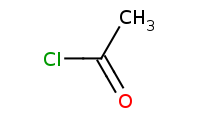
Note: Organometallic addition to a reactive carboxylic acid derivative such as this acid chloride. After the first addition, the C-Cl bond can break with a halide leaving group. The resulting ketone is even more reactive than the original acid chloride, thus a second equivalent of organometallic will add, yielding a tertiary alcohol. Refer to the [Mechanism] link for more details.
|
|
|
![[Li][Cu](C=C)C=C](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=%5BLi%5D%5BCu%5D%28C%3DC%29C%3DC&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
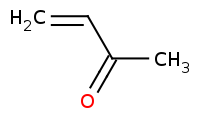
Note: Organocuprates are 'softer', less reactive carbon nucleophiles that will only add once to an acid chloride, yielding a ketone without any further reaction
|
|
|
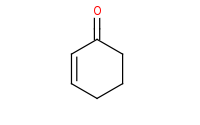
|
|
![C=C[C@@]1(CCCC=C1)O](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=C%3DC%5BC%40%40%5D1%28CCCC%3DC1%29O&width=200&height=125&arrowdesc=&extraImageSetting=amap)
Note: Nucleophiles can add to α,β unsaturated carbonyls directly at the carbonyl, or at the β carbon by conjugate addition. 'Hard', highly reactive nucleophiles like organolithiums which add irreversibly to carbonyls will tend to yield the direct 1,2 addition product.
![C=C[C@]1(CCCC=C1)O](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=C%3DC%5BC%40%5D1%28CCCC%3DC1%29O&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
|
|
|
|
![C=C[C@H]1CCCC(=O)C1](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=C%3DC%5BC%40H%5D1CCCC%28%3DO%29C1&width=200&height=125&arrowdesc=&extraImageSetting=amap)
Note: Less reactive, 'soft' nucleophiles like organocuprates which could add reversibly to the carbonyl or more likely to add by conjugate addition to α,β unsaturated carbonyls to yield a 1,4 addition product. After aqueous workup and respective tautomerization of an enol intermediate to keto form, the net result reproduces a carbonyl.
![C=C[C@@H]1CCCC(=O)C1](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=C%3DC%5BC%40%40H%5D1CCCC%28%3DO%29C1&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
|
![[C-]#N.[Na+]](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=%5BC-%5D%23N.%5BNa%2B%5D&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
C1)C#N](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=C1C%5BC%40H%5D%28CC%28%3DO%29C1%29C%23N&width=200&height=125&arrowdesc=&extraImageSetting=amap)
Note: Nitriles are another example of a 'soft' carbon nucleophile which add reversibly to carbonyls and will thus prefer to add by conjugate addition to α,β unsaturated carbonyls.
C1)C#N](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=C1C%5BC%40%40H%5D%28CC%28%3DO%29C1%29C%23N&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
|
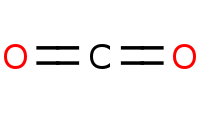
|
|
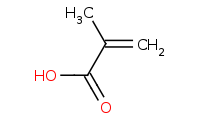
Note: Reactive, 'hard' organometallics like Grignard reagents have the unique ability to react with CO2 to yield carboxylic acids upon protonation by aqueous workup
|
|
|
![[Li]CCCCO[Si](C)(C)C](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=%5BLi%5DCCCCO%5BSi%5D%28C%29%28C%29C&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
(C)C)O](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=CC%28C%29%28CCCCO%5BSi%5D%28C%29%28C%29C%29O&width=200&height=125&arrowdesc=&extraImageSetting=amap)
Note: Silyl ether protecting groups will not interfere with organometallic reactivity, such as this addition to a ketone.
|
|
|
![C1COC(O1)CCC[Mg]Br](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=C1COC%28O1%29CCC%5BMg%5DBr&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
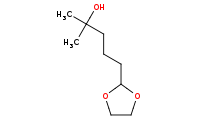
Note: Acetals will not interfere with organometallic reactivity, such as this ketone addition
|
|
|
![c1ccc(cc1)[Mg]Br](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=c1ccc%28cc1%29%5BMg%5DBr&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
![CC(=O)CCCO[Mg]Br](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=CC%28%3DO%29CCCO%5BMg%5DBr&width=200&height=125&arrowdesc=&extraImageSetting=amap)
ERROR: Organometallics act as strong bases and will deprotonate any slightly acidic protons before 'intended' reactivity like addition to a carbonyl occurs
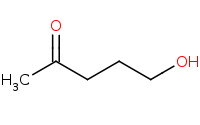
|
|
|
|
|
O](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=CC%28%3DO%29CC%5BC%40H%5D%28c1ccccc1%29O&width=200&height=125&arrowdesc=&extraImageSetting=amap)
Note: Aldehydes are more reactive than ketones based on lesser steric hindrance
O](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=CC%28%3DO%29CC%5BC%40%40H%5D%28c1ccccc1%29O&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
|
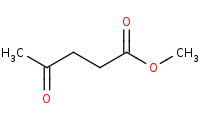
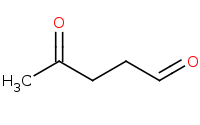
|
|
OC)(c1ccccc1)O](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=C%5BC%40%5D%28CCC%28%3DO%29OC%29%28c1ccccc1%29O&width=200&height=125&arrowdesc=&extraImageSetting=amap)
Note: Ketones are more reactive than esters based on the electron donating properties of the ester 'alcohol'
OC)(c1ccccc1)O](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=C%5BC%40%40%5D%28CCC%28%3DO%29OC%29%28c1ccccc1%29O&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
|
(C)C](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=CC%28%3DO%29CCCO%5BSi%5D%28C%29%28C%29C&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
(C)C)(c1ccccc1)O](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=C%5BC%40%5D%28CCCO%5BSi%5D%28C%29%28C%29C%29%28c1ccccc1%29O&width=200&height=125&arrowdesc=&extraImageSetting=amap)
Note: Organometallic reactivity, such as this addition to the carbonyl, is safe in the presence of the silyl ether protected alcohol
(C)C)(c1ccccc1)O](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=C%5BC%40%40%5D%28CCCO%5BSi%5D%28C%29%28C%29C%29%28c1ccccc1%29O&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
|
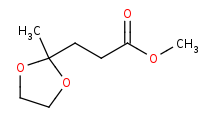
|
|
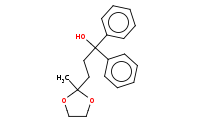
Note: Organometallic addition to the ester can proceed without affecting the acetal protected ketone. Note that 2 equivalents of organometallic reagent are added to carboxylic acid derivatives.
|
|
|
![[Li]c1ccccc1](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=%5BLi%5Dc1ccccc1&width=200&height=125&arrowdesc=&extraImageSetting=amap)
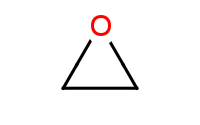
|
|
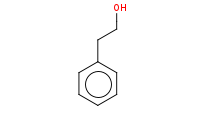
Note: Organolithiums act as highly reactive carbon nucleophiles which can open epoxides, yielding an alcohol after protonation by aqueous workup
|
|
|
![C[C@@H]1CO1](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=C%5BC%40%40H%5D1CO1&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
O](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=C%5BC%40H%5D%28Cc1ccccc1%29O&width=200&height=125&arrowdesc=&extraImageSetting=amap)
Note: Organolithiums open epoxides with an Sn2 mechanism which means attack will be favored at the less substituted site based on steric hindrance
|
|
|
![C/C=C/[Mg]Br](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=C/C%3DC/%5BMg%5DBr&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
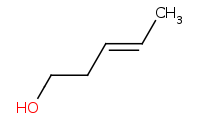
Note: Grignard organometallics also act as highly reactive carbon nucleophiles which can open epoxides in a convenient way to add a 2 carbon unit to a molecule
|
|
|
![C[C@H]1CO1](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=C%5BC%40H%5D1CO1&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
O](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=C/C%3DC/C%5BC%40H%5D%28C%29O&width=200&height=125&arrowdesc=&extraImageSetting=amap)
Note: Grignard organometallics also open epoxides by Sn2 mechanism with respective preference for less substituted site and stereospecific ring opening
|
|
|
![[Li]/C=C/C](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=%5BLi%5D/C%3DC/C&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
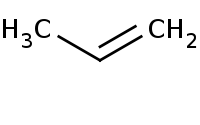
Warning: Organometallics act as very strong bases and thus must not be used in the presence of any protic solvent or even slightly acidic protons or else a simple acid-base reaction will result that is usually unproductive
![[Li+].CC[O-]](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=%5BLi%2B%5D.CC%5BO-%5D&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
|
|
|
|
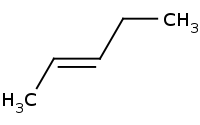
ERROR: Even though Grignard reagents and organolithium compounds generally act as strong carbon nucleophiles, contrary to intuition, they cannot reliably be used in Sn2 reactions against alkyl halides
|
|
|
|
|
|
O](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=C%5BC%40H%5D%28C%23N%29O&width=200&height=125&arrowdesc=&extraImageSetting=amap)
Note: Nucleophilic addition of a cyanide ion to a carbonyl to form a cyanohydrin. Reaction pH must be carefully controlled to manage reversible reaction
O](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=C%5BC%40%40H%5D%28C%23N%29O&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
|
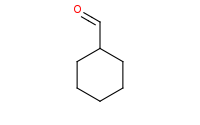
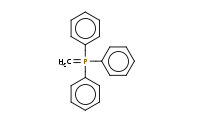
|
|
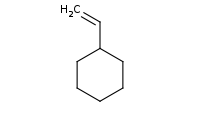
Note: The Wittig reagent reacts with aldehydes to replace the C=O bond with a new C=C double bond
|
|
|
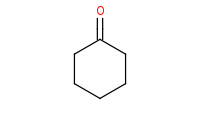
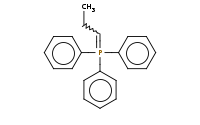
|
|
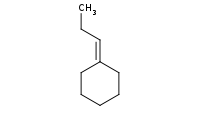
Note: The Wittig reagent reacts with ketones to replace the C=O bond with a new C=C double bond
|
|
|
|
|
Note: Amide formation from a reactive acid chloride. An extra equivalent of amine or other base is necessary to soak up protons that are produced in the reaction.
|
|
|
|
|
![CCNC(=O)C.CC(=O)[O-]](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=CCNC%28%3DO%29C.CC%28%3DO%29%5BO-%5D&width=200&height=125&arrowdesc=&extraImageSetting=amap)
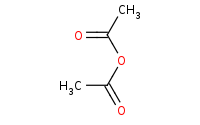
Note: Amide formation from a reactive anhydride. An extra equivalent of amine or other base is necessary to soak up protons that are produced in the reaction.
|
|
|
|
|
![CC[NH3+].CC(=O)[O-]](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=CC%5BNH3%2B%5D.CC%28%3DO%29%5BO-%5D&width=200&height=125&arrowdesc=&extraImageSetting=amap)
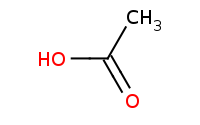
Warning: Amide preparation from a carboxylic acid will not work directly because acid-base reactions will occur first.
|
|
|
|
|
|
Note: Amide preparation from an ester is viable, though less reactive than using an acid chloride or anhydride.
|
|
|
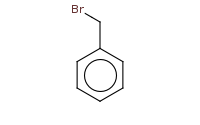
|
|
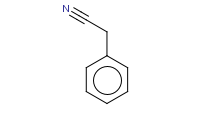
Note: The most general way to prepare nitriles is to simply perform an Sn2 substitution with a nitrile ion against an alkyl halide.
|
|
|
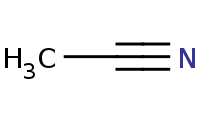
|
|
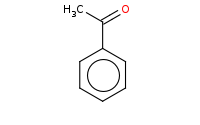
Note: Organometallic reagents can add to nitriles to yield a primary (metallo) imine that will be hydrolyzed to a ketone upon aqueous workup.

|
|
|
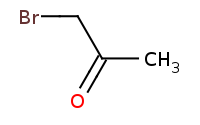
|
|
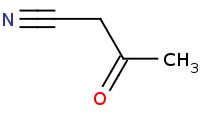
Note: Alpha halogenation of carbonyls has the effect of introducing an electrophile at the alpha position. In select cases, this is convenient for substituting a nucleophile at the alpha position.
|
|
|
![[Li+].CCC(=C)[O-]](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=%5BLi%2B%5D.CCC%28%3DC%29%5BO-%5D&width=200&height=125&arrowdesc=&extraImageSetting=amap)
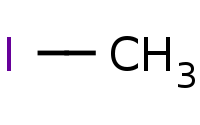
|
|
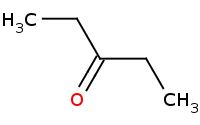
Note: Direct enolate alkylation. Preparation of an enolate has the effect of giving the alpha carbon the character of a nucleophile, which can then be used to substitute against unhindered (methyl or primary) alkyl halides.
|
|
|
![C/C(=C/C(=O)C)/[O-].[Na+]](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=C/C%28%3DC/C%28%3DO%29C%29/%5BO-%5D.%5BNa%2B%5D&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
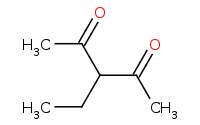
Note: Double activated enolates can be directly alklyated effectively as well.
|
|
|
![CC/C(=C(\C)/[O-])/C(=O)C.[Na+]](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=CC/C%28%3DC%28%5CC%29/%5BO-%5D%29/C%28%3DO%29C.%5BNa%2B%5D&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
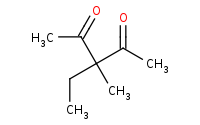
Note: A second alkylation of the double activated enolate.
|
|
|
|
|
|
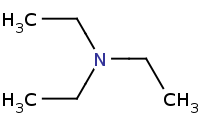
Warning: Amines (and ammonia) are good nucleophiles, but are not generally useful for preparing primary amines because of rampant over-alkylation.
|
|
|
![c1ccc2c(c1)C(=NC2=O)[O-].[Na+]](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=c1ccc2c%28c1%29C%28%3DNC2%3DO%29%5BO-%5D.%5BNa%2B%5D&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
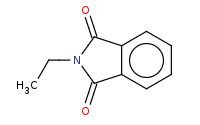
Note: Gabriel synthesis of primary amines includes a typical Sn2 reaction against an unhindered alkyl halide, using the deprotonated phthalimide as the nitrogen nucleophile. Note that further (over) alkylation is not a problem for this product.
|
|
|
![c1ccc(cc1)[N+]#N.[Br-]](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=c1ccc%28cc1%29%5BN%2B%5D%23N.%5BBr-%5D&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
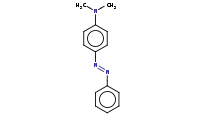
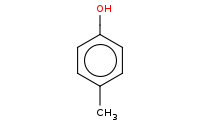
Note: Arenediazonium compounds are decent electrophiles and can react with strongly activated benzene derivatives (NH2, NHR, NR2 or OH substituents) in an EAS (electrophilic aromatic substitution) coupling reaction to yield a highly conjugated azo dye.
|
|
|
|
|
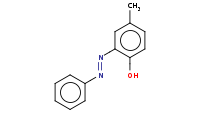
Note: Azo coupling of arenediazonium compounds generally yields the para isomer, but ortho isomers can also be produced if the para position is blocked.
|
|
|
|
|
|
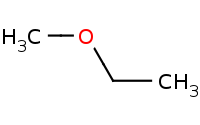
Note: Sn2 - Primary Alkyl Halide, Strong Nucleophile
|
|
|
Br](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=CC%5BC%40%40H%5D%28C%29Br&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
OC](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=CC%5BC%40H%5D%28C%29OC&width=200&height=125&arrowdesc=&extraImageSetting=amap)
Note: E2, Sn2 Competition - Secondary Alkyl Halide, Strong Nucleophile
Note stereospecific inversion of Sn2
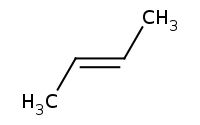
Note: Zaitsev rule preference for more highly substituted alkene and trans substituents
|
|
|
(C(C)C)Br](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=CC%5BC%40%40%5D%28C%29%28C%28C%29C%29Br&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
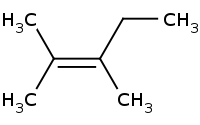
Note: E2 - Tertiary Alkyl Halide, Strong Nucleophile
Note Zaitsev rule preference for more highly substituted alkene
|
|
|
|
|
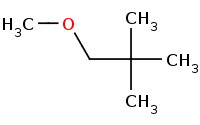
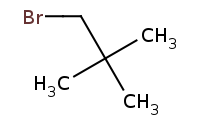
DISFAVORED: No reaction! Primary alkyl halide, but extreme beta-branching (neo-pentyl group) is too stericly crowded for Sn2 and no beta-Hydrogens exist for E2
|
|
|
[C@@H](C)Br](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=CC%5BC%40%40H%5D%28C%29%5BC%40%40H%5D%28C%29Br&width=200&height=125&arrowdesc=&extraImageSetting=amap)
![CC(C)(C)[O-].[Na+]](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=CC%28C%29%28C%29%5BO-%5D.%5BNa%2B%5D&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
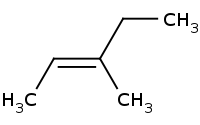
Note: Stereospecific E2 requires proton and leaving group to be anti-periplanar
|
|
|
[C@H](C)Br](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=CC%5BC%40%40H%5D%28C%29%5BC%40H%5D%28C%29Br&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
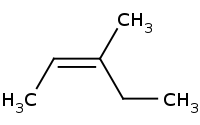
Note: Stereospecific E2 requires proton and leaving group to be anti-periplanar, even though this results in the largest substituents being cis to each other
|
|
|
![CC[C@@H]1CCC[C@H]([C@H]1Br)C](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=CC%5BC%40%40H%5D1CCC%5BC%40H%5D%28%5BC%40H%5D1Br%29C&width=200&height=125&arrowdesc=&extraImageSetting=amap)
|
|
![CC[C@@H]1CCCC(=C1)C](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=CC%5BC%40%40H%5D1CCCC%28%3DC1%29C&width=200&height=125&arrowdesc=&extraImageSetting=amap)
Note: Stereospecific E2 requires proton and leaving group to be anti-periplanar. Within ring system, this means they must assume a trans-diaxial conformation
|
|
|
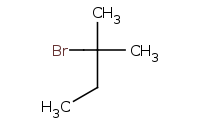
|
|
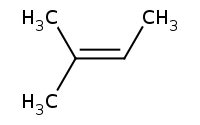
Note: E2 elimination as usual, Zaitsev rule preference for more highly substituted alkene
|
|
|
|
|
|
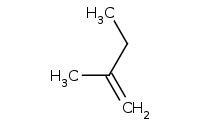
Caution: Bulky base AND bulky alkyl halide. Anti-Zaitsev preference for LEAST substituted alkene
|
|
|
|
|
|
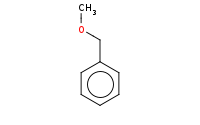
Note: Sn2 at primary benzylic halide
|
|
|
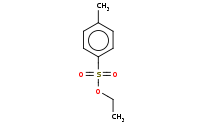
|
|
Note: Tosylate acts as a leaving group comparable to Cl- or Br- in this Sn2 reaction
|
|
|
|
|
|
O](https://re.edugen.wiley.com/arrow-webapp/ArrowWebService?action=smi2png&smiles=C%5BC%40%40H%5D%28COC%29O&width=200&height=125&arrowdesc=&extraImageSetting=amap)
Note: Base-driven epoxide opening. Similar to Sn2, prefer less sterically hindered end
|
|
|
|
|
|
(0.081 sec)
Link
|
|